
What Is Phelan-McDermid Syndrome? Causes, Symptoms & Treatment
Phelan-McDermid syndrome is a rare genetic disorder that affects how a person’s brain develops and functions, and how the body functions. This guide provides a comprehensive overview of Phelan-McDermid syndrome, including causes, symptoms and treatment. Click on any of the links below to navigate directly to the answers you need:
What Is Phelan-McDermid Syndrome?
Phelan-McDermid syndrome (pronounced FEY-luhn mick-DUR-mid) is a rare genetic disorder involving chromosome 22 that can affect many critical functions in a person's body — from learning and communicating to eating and sleeping.
Children with this complex medical condition are born with a genetic difference on their 22nd chromosome that affects brain development and functioning. This results in intellectual and physical disabilities that vary from person to person. Some people with Phelan-McDermid syndrome lose essential skills, and most require long-term care and medical attention.
You may see and hear Phelan-McDermid syndrome abbreviated as PMS. You may also see Phelan-McDermid syndrome referred to as 22q13 deletion syndrome, based on the location of the genetic change in the body.
What causes Phelan-McDermid syndrome?
Phelan-McDermid syndrome occurs when a person is born with a genetic deletion or change (variant) on their 22nd chromosome.
Chromosomes are the building blocks of the human body. Each cell contains 23 pairs of chromosomes that carry our genes. Chromosome 22 contains hundreds of genes that provide instructions for making proteins that perform various roles in the body. When a piece of DNA is missing on the chromosome, or one of those genes doesn’t work the way it should, it can lead to altered brain development and functioning, as is the case with Phelan-McDermid syndrome.
You’ll hear medical experts describe Phelan-McDermid syndrome’s causes as one of the following:
A deletion or other structural difference on the end of the long arm of chromosome 22, in a region known as 22q13
A disease-causing (pathogenic) variant in the SHANK3 gene, which is a change to a single gene on chromosome 22 that’s involved in the functioning of connections between brain cells
Researchers are trying to understand if there are differences in symptoms between people who have a deletion or genetic change to chromosome 22 and those with a change to their SHANK3 gene.
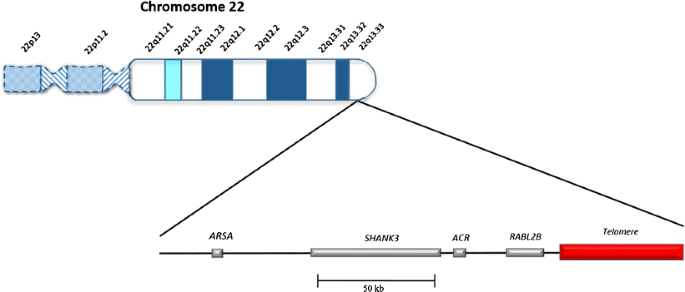
What does 22q13 mean?
Geneticists use a combination of numbers and letters to describe the location of genes on a chromosome. The deletion or genetic change involved in Phelan-McDermid syndrome is at location 22q13. Sometimes you’ll even hear Phelan-McDermid syndrome called 22q13 deletion syndrome (pronounced twenty-two Q one three). This means that it is located on chromosome 22 on the longer of the two arms of the chromosome (arm q) and position 13 on that long arm.
What is the Shank3 Gene and What Does a Shank3 Mutation Mean?
SHANK3 is a gene that is important for the development and function of the nervous system. Genes are like instruction manuals in our bodies that help cells grow and work. SHANK3 is one such gene that tells our bodies how to make a protein also called SHANK3. Proteins carry out many of the essential functions in our cells.
The SHANK3 protein helps nerve cells, or neurons, talk to each other. This communication is needed for many brain functions, such as learning, memory, and social interactions. It is also needed for the signals the brain sends to the rest of the body, for functions like walking.
How does SHANK3 do this? It can be thought of as a support structure. It provides the structure necessary for neurons to receive signals. Read more.
Is Phelan-McDermid syndrome inherited?
For most people born with Phelan-McDermid syndrome, their genetic change happens randomly or sporadically (de novo) when the egg and sperm are forming or during fetal development. In a lower percentage of cases, a parent may carry a genetic change that does not affect them but could increase the chances of PMS in a child. Genetic testing of the parents and child can help determine this. In any case, it is not the parent’s fault or something the parent did. Phelan-McDermid syndrome is linked to errors that occur when DNA is copied.
What are the signs and symptoms of Phelan-McDermid syndrome?
Phelan-McDermid syndrome can cause a wide range of medical, intellectual, and behavioral challenges that vary in severity from person to person based on the size and severity of their genetic change.
Although not present in every case, the most common symptoms of Phelan-McDermid syndrome are low or weak muscle tone (hypotonia), developmental delays and absent or delayed speech.
People with Phelan-McDermid syndrome often show symptoms very early in childhood, sometimes at birth and within the first six months of life. Some people are diagnosed later in life, even in adulthood, after new symptoms arise or new testing is done. Developmental delays associated with the syndrome include not achieving typical milestones on time, like rolling over, sitting up, walking, or talking.
Other common symptoms of Phelan-McDermid syndrome include:
- Moderate to severe intellectual disabilities
- Speech delays or problems
- Low or weak muscle tone (hypotonia)
- Sleep disturbance
- Poor feeding
- Difficulties with toilet training and chronic constipation
- Seizures – about 40% of people develop seizures that range from mild to severe
- Behavioral challenges
- Decreased perspiration and heat intolerance, and lymphedema
75%
of people with Phelan-McDermid syndrome have been diagnosed with an Autism Spectrum Disorder. Behavioral issues may stem from autism (e.g., repetitive behaviors), poor communication skills, or an unknown origin.
Common Medical Issues Associated With Phelan-McDermid Syndrome
Fluid-filled sacs that can occur on the surface of the brain. They occur in over 15% of individuals with 22q13 deletion compared to about 1% in the general population. Most arachnoid cysts are present early in life and may remain asymptomatic for many years. Enlargement of the cyst can cause increased pressure on the brain and lead to symptoms such as incessant crying bouts, irritability, severe headaches, cyclic vomiting, or seizures. Brain imagining studies, such as MRI, should be performed if a cyst is suspected.
Due to low muscle tone, individuals may have swallowing difficulties that could result in the aspiration of fluids/solids into the airways. Simple aspiration could lead to aspiration pneumonia and cause lung damage. If your child exhibits coughing while swallowing thin liquids and/or solids or has frequent lower respiratory infections, a gastroenterologist and/or pulmonologist should be consulted to determine what treatments are necessary. In severe cases, a feeding tube (GI tube) may be necessary to reduce the risk of aspiration.
Your child may be diagnosed with autism or show some of the features of autism. These include deficits in social communication, difficulty making eye contact, repetitive behavior, and limited or restricted interests. It is not surprising that a child with delayed or absent speech has difficulty in social communication (See speech delays/absent speech). Estimates of the prevalence of autism in PMS have been as high as 84%.
Chronic diarrhea refers to loose, watery stools that persist for 4 weeks or more. Depending on the cause of diarrhea, it may be controlled with a special diet. An individual with diarrhea should be closely monitored to ensure that they do not become dehydrated. Persistent diarrhea may require treatment by a gastroenterologist.
With PMS, your child may not meet milestones for various physical and mental activities. Your child may require continual individualized therapies such as physical therapy (PT), occupational therapy (OT), and speech therapy (ST), and may benefit from other therapies as well, such as hippo/horse therapy, music therapy, and water therapy.
The contents of the stomach may flow back up into the esophagus because of a problem with the muscles in the lower esophagus. If not treated, this could cause severe vomiting and damage to the esophagus. Individuals should be treated by a gastroenterologist and may require medication or other procedures to control reflux.
The birth weight, length, and head size are usually appropriate for gestational age. In general, growth is normal. In childhood and adolescence, the height is usually normal but may be advanced for age. Weight may not be increased, so your child may be described as “tall and thin”. The adult height is usually within the normal range. Head size is variable and may be normal, large, or small for age.
This is a hallmark feature of the syndrome and is usually the first presenting feature observed in the newborn. The decreased tone of skeletal muscles is characterized by weakness/floppiness. It is often associated with difficulty feeding and swallowing, weak cry, and poor head control. Hypotonia should be evaluated by a neurologist.
The swelling of a body part is caused by the abnormal accumulation of lymph fluid in the limbs. This often occurs in the teenage years. Mild cases can be helped by elevation of the affected limb and use of compression stockings. Severe cases may require evaluation by a vascular surgeon.
The ventricles of the brain may be enlarged due to the accumulation of cerebrospinal fluid or because there is less tissue around the ventricle due to delayed brain growth. Mildly enlarged ventricles without other complications may resolve on their own. Cases in which fluid continues to accumulate may require a shunt to reduce the pressure on the brain. As a precaution, individuals should be under the care of a neurologist who may order brain imaging, such as an MRI. Other neurological problems may include reduced myelination, frontal lobe hypoplasia, agenesis of the corpus callosum, and seizures.
Otitis media is inflammation of the middle ear, commonly called an ear infection. Individuals with recurrent ear infections may require medications or insertion of ear tubes by an Ear, Nose and Throat (ENT) specialist.
The type and severity of seizures are variable in PMS. There are mild seizures in which the person has staring spells or “tunes out” but does not have any physical spasms. Other individuals may experience full seizures with loss of consciousness, jerky movements, and stiffening of the joints. These seizures are usually controlled with medication and the affected individual should be under the care of a neurologist or epileptologist.
One of the most common features of individuals with the syndrome is delayed or absent speech. Individuals will benefit from speech therapy and may be able to communicate using sign language or some type of augmentative communication device depending on their cognitive skills and fine motor capabilities. The receptive language is typically better than the expressive language so a child may understand what is said but cannot respond verbally.
Individuals who are immobile may have problems with a subluxing hip joint where the femur bone does not grow properly into the hip joint. These individuals should be under the care of an orthopedist and may require further treatments.
Normally, urine flows from the kidneys through the ureters to the bladder. VUR is an abnormal flow of urine from the bladder, back up the ureter, and back into the kidneys. It is recommended that a renal ultrasound be performed as soon as possible after an individual has been diagnosed with PMS to determine if VUR or other asymptomatic renal problems are present. Individuals with renal anomalies should be monitored by a urologist.
Drooping of the upper eyelid (ptosis) is seen in individuals with this syndrome. The eyes may look partially shut as if they are ready to fall asleep. Wandering eye is referred to as strabismus and may require treatment by an ophthalmologist. Vision difficulties can result in the extensive use of peripheral vision and in poor depth perception.
Common Physical and Facial Features Associated With Phelan-McDermid Syndrome
In addition to medical issues described above, individuals with Phelan-McDermid syndrome may have the following physical or facial features:
This means having a long skull with a prominent forehead – the head can be measured to determine if your child is dolichocephalic. You may be able to look at your child’s head shape and recognize that it is longer than usual for his/her age group.
The eye openings (the space between our eyelids) are down-slanted, which means that the outer corner of the eye is lower than the inner corner. In contrast, individuals with PMS may have up slanted palpebral fissures, so the eyes slant up.
The ears may be abnormally developed or poorly formed. The external part of the ear may not be folded as much as usual. In addition to being dysplastic, the ears may be large.
Often the toenails are thin, flaky, and peel off easily in the infant and young child. Many parents say they never have to cut their child’s toenails because they flake off spontaneously. As the child gets older, the toenails may become hard and brittle.
The vertical fold of skin on the inner corner of the eye (on each side of the nose) is excessive and covers the inner corner of the eye. This feature may be seen in individuals with PMS.
The roof of the mouth is higher than usual. A high arched palate is considered a normal variant in the general population but is seen more frequently in PMS.
The head may be largely due to an unidentified cause or due to a known cause, such as enlarged ventricles. Macrocephaly has been reported in about 25% of individuals with the syndrome.
This means that the head is small for age and is more common in individuals with short stature. Microcephaly occurs in fewer than 15% of individuals with the syndrome.
This refers to minor facial anomalies that occur fairly frequently and do not pose a significant health risk. The features include ptosis, epicanthal folds, and puffiness around the eyes, puffy cheeks, long thick eyelashes, and down-slanting palpebral fissures. The presence of two or more minor anomalies may prompt a search for major defects and may aid in the diagnosis of a particular syndrome.
A single crease (line) along the middle of the palm of the hand is sometimes seen in PMS. Usually, there are three major creases on the palm: a crease that goes from the pinkie side of the palm and curves up toward the index finger, another crease that starts on the pinkie side of the hand and goes between the index finger and the thumb and the third crease that goes from above the thumb toward the wrist. In individuals with a single palmar crease, there is a straight line across the palm. A single palmar crease is fairly common in the general population occurring in about 1 in 30 individuals.
Webbing or an extra fold of skin that usually occurs between the second and third toes. This is a fairly common variant in the general population and may be inherited from one parent, unrelated to the syndrome.
How common is Phelan-McDermid syndrome?
Phelan-McDermid syndrome is considered a rare disease. It is estimated that approximately 1 in 10,000 people may have this genetic condition. This is based on scientific studies that look at the overlap of Phelan-McDermid syndrome with autism.
Because Phelan-McDermid syndrome can be challenging to detect and diagnose, and because many medical professionals are still unfamiliar with the condition, it is likely underdiagnosed. The Phelan-McDermid Syndrome Foundation has more than 3,100 members with a reported diagnosis, the largest Phelan-McDermid community in the world that is growing at a steady rate of almost one new person every other day.
How is Phelan-McDermid syndrome diagnosed?
Anyone exhibiting the following signs or symptoms should be tested for a Phelan-McDermid syndrome diagnosis:
- Intellectual disability, with or without autism
- Atypical physical features
- Severe speech delay or a history of neonatal hypotonia (low muscle tone) of unknown cause or regression
Your medical professional will order the appropriate genetic diagnostic tests.
What tests are used to diagnose Phelan-McDermid syndrome?
Three types of tests are most commonly used to diagnose Phelan-McDermid syndrome. These include 1) chromosomal microarray analysis, 2) chromosome structural tests, known as FISH or karyotype, and 3) sequencing. This section explores each of these types of tests to diagnose Phelan-McDermid syndrome.
Chromosomal Microarray Analysis (CMA) is the genetic test most commonly used to diagnose Phelan-McDermid syndrome. It involves providing a small amount of blood. This technology can detect chromosomal deletions or duplications. However, these tests cannot “see” the structure of the chromosome, which can help reveal additional risks when a person is found to have a 22q13 deletion specifically.
If CMA testing identifies a 22q13 deletion, what additional risks should you consider for testing?
There are two possibilities for which chromosome structural tests are ordered if a 22q13 deletion is identified through CMA.
First, a small subset of people with Phelan-McDermid syndrome has an unusual formation of their 22nd chromosome into a ring shape (Ring chromosome 22). This is caused by a deletion on both ends of the chromosome. Ring chromosome 22 carries some additional medical concerns to be monitored over time.
Second, some people diagnosed with a deletion may have an unusual rearrangement (translocation) where genetic material has been shuffled between chromosomes. This can be more likely based on a parent’s genetics, in which case the parent should receive genetic testing to learn of risks to future children.
In either of these cases, chromosome structural tests should be ordered.
Chromosomal structural tests may detect larger deletions and visualize the structure of the chromosome. These tests are called fluorescence in situ hybridization (FISH) or conventional chromosome analysis (karyotype). This type of test is necessary to identify translocations (unusual rearrangements) or ring chromosomes (an unusual formation of a chromosome into a ring shape). These tests can also be used to study parents of individuals with Phelan-McDermid syndrome to determine if either parent carries a genetic difference in their 22nd chromosome that could affect their child.
Sequencing is a way of reading the genome at a very high resolution to report exactly what genes, and versions of genes, are present. It can help detect changes to a single gene which can result in a Phelan-McDermid syndrome diagnosis, even if a deletion is not found with other testing methods.
Whole Exome Sequencing (WES) focuses on the genes that are involved in making proteins, which are responsible for many of the functions in our cells.
Whole Genome Sequencing (WGS) will read every single gene in the genome, including those involved in regulating other genes, and is the most complete sequencing method. Because sequencing methods read every “base” or letter of these genes, they can be used to detect very small mutations (alterations), such as in the SHANK3 gene. These tests can also provide detailed information for the scientific and medical study of which genes matter in Phelan-McDermid syndrome.
Please refer to the diagram below to help you and your doctor decide if follow-up testing is needed.
How is Phelan-McDermid syndrome treated?
Currently, there are no treatments or cures for the underlying cause of Phelan-McDermid syndrome – something the Phelan-McDermid Syndrome Foundation works every minute of every day to change.
People with Phelan-McDermid syndrome receive treatment based on the symptoms they experience. Most individuals with the condition need a team of medical, developmental and educational specialists to address symptoms and challenges, such as challenging behaviors, seizures, sleep disturbance, hypotonia, poor feeding, chronic constipation, gastroesophageal reflux, renal problems, decreased perspiration and heat intolerance, and lymphedema.
Learn about the research the Phelan-McDermid Syndrome Foundation is leading to deliver treatments and cures faster.
What is the life expectancy for people with Phelan-McDermid Syndrome?
Life expectancy for individuals with Phelan-McDermid syndrome is uncertain and will vary from person to person. While the condition does not typically cause life-threatening issues, people experience lifelong challenges and complications that can significantly impact an individual and their family’s quality of life.
The Phelan-McDermid Syndrome Foundation is doing everything it takes to make today better and the future brighter for every person living with Phelan-McDermid syndrome – from the moment of diagnosis to the delivery of treatments and cures. We will not stop until Phelan-McDermid syndrome is treatable and curable and every person and family affected is thriving.
Click on any of the links below to navigate directly to the answers you need:
- What is Phelan-McDermid syndrome?
- What causes Phelan-McDermid syndrome?
- What are the signs and symptoms of Phelan-McDermid syndrome?
- How is Phelan-McDermid syndrome diagnosed?
- How common is Phelan-McDermid syndrome?
- How is Phelan-McDermid syndrome treated?
- What is the life expectancy for someone with Phelan-McDermid syndrome?

All scientific and medical content from the Phelan-McDermid Syndrome Foundation is carefully created and/or reviewed by Kate Still, Ph.D., scientific director for the Phelan-McDermid Syndrome Foundation, chair of its Scientific Advisory Committee and principal investigator for the PMS DataHub. She works closely with members of the organization’s medical and scientific advisory committees to bring families and medical professionals the information they need in a way they can understand and act on it.
All scientific and medical content from the Phelan-McDermid Syndrome Foundation is carefully created and/or reviewed by Kate Still, Ph.D., scientific director for the Phelan-McDermid Syndrome Foundation, chair of its Scientific Advisory Committee and principal investigator for the PMS DataHub. She works closely with members of the organization’s medical and scientific advisory committees to bring families and medical professionals the information they need in a way they can understand and act on it.